





Aldone al teknologio, la sintezo de glikozidoj ĉiam interesis sciencon, ĉar ĝi estas tre ofta reakcio en la naturo. Lastatempaj artikoloj de Schmidt kaj Toshima kaj Tatsuta, same kiel multaj referencoj cititaj tie, komentis pri vasta gamo da sintezaj potencialoj.
En la sintezo de glikozidoj, plursukeraj komponantoj estas kombinitaj kun nukleofiloj, kiel alkoholoj, karbonhidratoj aŭ proteinoj. Se necesas selektiva reakcio kun unu el la hidroksilaj grupoj de karbonhidrato, ĉiuj aliaj funkcioj devas esti protektitaj en la unua paŝo. Principe, enzimaj aŭ mikrobaj procezoj, pro sia selektiveco, povas anstataŭigi kompleksajn kemiajn protektajn kaj deprotektajn paŝojn por selekte forigi glikozidojn en regionoj. Tamen, pro la longa historio de alkilaj glikozidoj, la apliko de enzimoj en la sintezo de glikozidoj ne estis vaste studita kaj aplikita.
Pro la kapacito de taŭgaj enzimsistemoj kaj altaj produktokostoj, enzima sintezo de alkilaj poliglikozidoj ankoraŭ ne estas preta por esti ĝisdatigita al la industria nivelo, kaj kemiaj metodoj estas preferataj.
En 1870, MAcolley raportis la sintezon de "acetoklorhidrozo" (1, figuro 2) per reakcio de dekstrozo (glukozo) kun acetilklorido, kio fine kondukis al la historio de glikozidaj sintezaj vojoj.
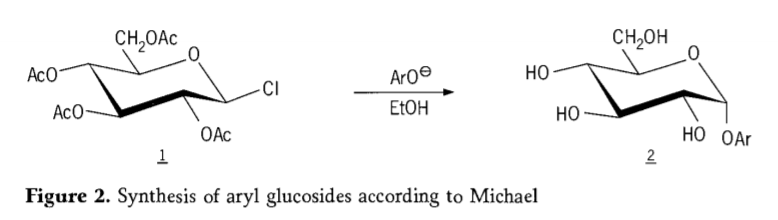
Tetra-O-acetil-glukopiranosilaj halogenidoj (acetohaloglukozoj) poste montriĝis utilaj intermediatoj por la stereoselektiva sintezo de puraj alkilaj glukozidoj. En 1879, Arthur Michael sukcesis prepari definitivajn, kristaligeblajn arilajn glikozidojn el la intermediatoj kaj fenolatoj de Colley. (Aro-, Figuro 2).
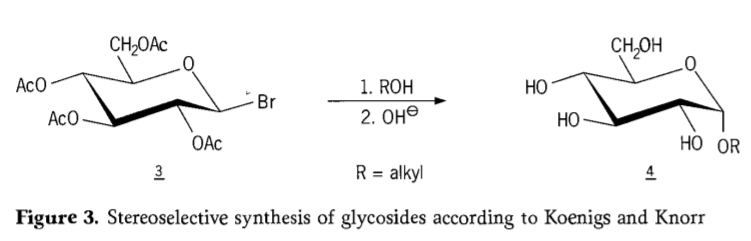
En 1901, la sintezo de Michael por larĝa gamo de karbonhidratoj kaj hidroksilaj aglikonoj, kiam W. Koenigs kaj E. Knorr enkondukis sian plibonigitan stereoselektivan glikosidigan procezon (Figuro 3). La reakcio implikas SN2-anstataŭigon ĉe la anomera karbono kaj okazas stereoselektive kun inversio de konfiguracio, produktante ekzemple la α-glukozidon 4 el la β-anomero de la aceobromoglukoza intermediato 3. La Koenigs-Knorr-sintezo okazas en la ĉeesto de arĝentaj aŭ hidrargaj promotoroj.
En 1893, Emil Fischer proponis principe malsaman aliron al la sintezo de alkilaj glukozidoj. Ĉi tiu procezo nun estas bone konata kiel la "Fischer-glikosidiĝo" kaj konsistas el acido-katalizita reakcio de glikozoj kun alkoholoj. Tamen, ĉiu historia raporto ankaŭ devus inkluzivi la unuan raportitan provon de A. Gautier en 1874, konverti dekstrozon per anhidra etanolo en la ĉeesto de klorida acido. Pro misgvida elementa analizo, Gautier kredis, ke li akiris "diglukozon". Fischer poste pruvis, ke la "diglukozo" de Gautier estis fakte ĉefe etila glukozido (Figuro 4).
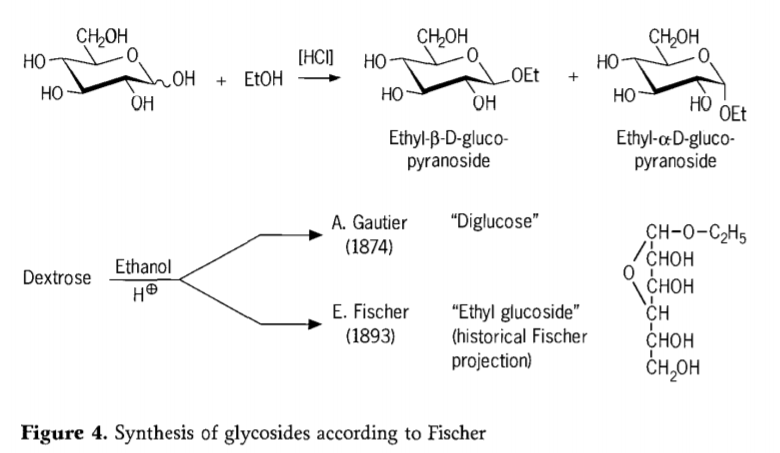
Fischer ĝuste difinis la strukturon de etila glukozido, kiel videblas el la historia furanozida formulo proponita. Fakte, la glikozidaj produktoj de Fischer estas kompleksaj, plejparte ekvilibraj miksaĵoj de α/β-anomeroj kaj piranozidaj/furanozidaj izomeroj, kiuj ankaŭ konsistas el hazarde ligitaj glikozidaj oligomeroj.
Sekve, individuaj molekulaj specioj ne estas facile izoleblaj el Fischer-reakciaj miksaĵoj, kio estis grava problemo en la pasinteco. Post iom da plibonigo de ĉi tiu sintezmetodo, Fischer poste adoptis la Koenigs-Knorr-sintezon por siaj esploroj. Uzante ĉi tiun procezon, E. Fischer kaj B. Helferich estis la unuaj, kiuj raportis la sintezon de longĉena alkilglukozido montranta surfaktantajn ecojn en 1911.
Jam en 1893, Fischer ĝuste rimarkis esencajn ecojn de alkilaj glikozidoj, kiel ekzemple ilian altan stabilecon rilate al oksidado kaj hidrolizo, precipe en forte alkalaj medioj. Ambaŭ karakterizaĵoj estas valoraj por alkilaj poliglikozidoj en aplikoj de surfaktantaĵoj.
Esplorado rilata al la glikosidiga reakcio ankoraŭ daŭras kaj pluraj interesaj vojoj al glikozidoj estis evoluigitaj en la lastatempa pasinteco. Kelkaj el la proceduroj por la sintezo de glikozidoj estas resumitaj en Figuro 5.
Ĝenerale, kemiaj glikosidigprocezoj povas esti dividitaj en procezojn kondukantajn al kompleksaj oligomeraj ekvilibroj en acid-katalizita glikozilinterŝanĝo.
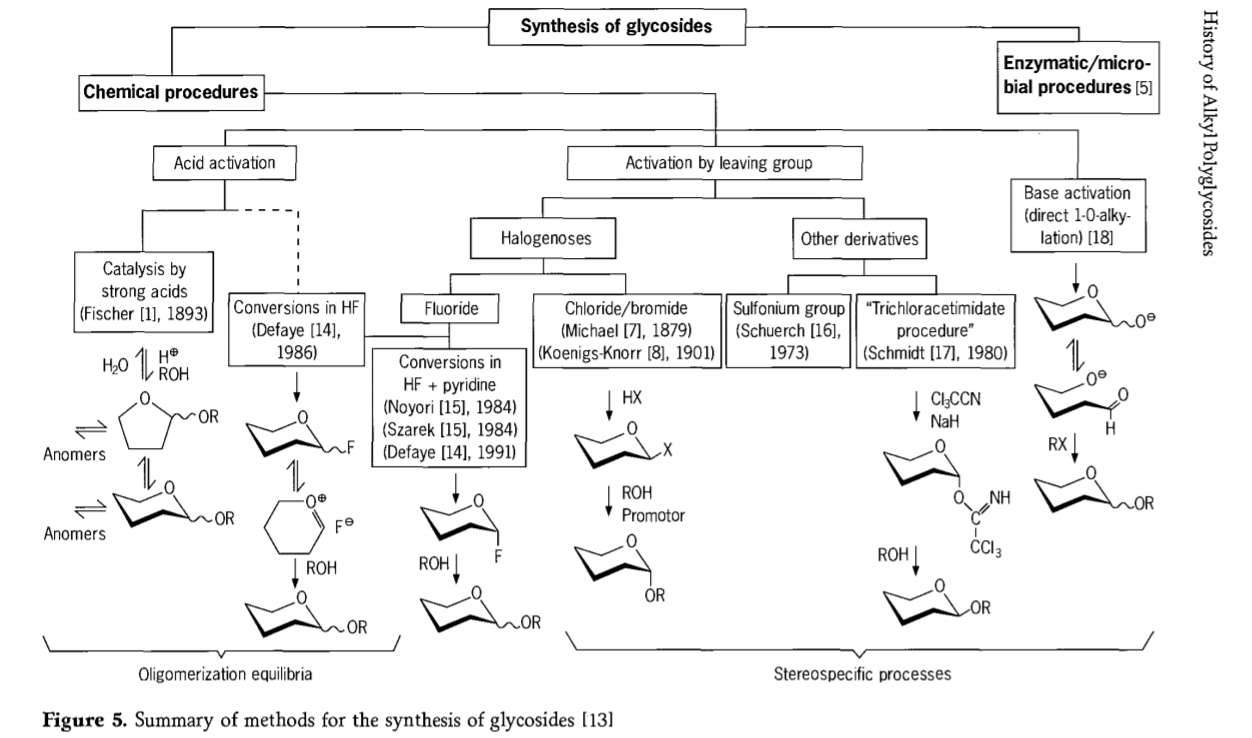
Reagoj sur konvene aktivigitaj karbonhidrataj substratoj (glikozidaj reakcioj de Fischer kaj hidrogenfluoridaj (HF) reakcioj kun neprotektitaj karbonhidrataj molekuloj) kaj kinetike kontrolitaj, nemaligeblaj kaj ĉefe stereotaksaj anstataŭigaj reakcioj. Dua tipo de proceduro povas konduki al la formado de individuaj specioj anstataŭ kompleksaj miksaĵoj de reakcioj, precipe kiam kombinite kun konservadaj grupaj teknikoj. Karbonhidratoj povas lasi grupojn sur la ektopa karbono, kiel ekzemple halogenaj atomoj, sulfoniloj aŭ trikloroacetimidataj grupoj, aŭ esti aktivigitaj de bazoj antaŭ konvertiĝo al triflataj esteroj.
En la aparta kazo de glikosidigoj en hidrogena fluorido aŭ en miksaĵoj de hidrogena fluorido kaj piridino (piridinia poli[hidrogena fluorido]), glikozilaj fluoridoj formiĝas surloke kaj glate konvertiĝas en glikozidojn, ekzemple kun alkoholoj. Hidrogena fluorido montriĝis esti forte aktiviga, nedegradanta reakcia medio; ekvilibra aŭtokondensiĝo (oligomerigo) estas observata simile al la Fischer-procezo, kvankam la reakcia mekanismo estas verŝajne malsama.
Kemie puraj alkilaj glikozidoj taŭgas nur por tre specialaj aplikoj. Ekzemple, alkilaj glikozidoj estis sukcese uzitaj en biokemia esplorado por la kristaliĝo de membranaj proteinoj, kiel ekzemple la tridimensia kristaliĝo de porino kaj bakteriorhodopsino en ĉeesto de oktila β-D-glukopiranosido (pliaj eksperimentoj bazitaj sur ĉi tiu laboro kondukis al la Nobel-premio pri kemio por Deisenhofer, Huber kaj Michel en 1988).
Dum la disvolviĝo de alkilaj poliglikozidoj, stereoselektivaj metodoj estis uzitaj je laboratorioskalo por sintezi diversajn modelajn substancojn kaj studi iliajn fizik-kemiajn ecojn. Pro ilia komplekseco, la malstabileco de intermediatoj kaj la kvanto kaj kritika naturo de procezaj rubaĵoj, sintezoj de la tipo Koenigs-Knorr kaj aliaj protektaj grupaj teknikoj kreus signifajn teknikajn kaj ekonomiajn problemojn. Fischer-tipaj procezoj estas kompare malpli komplikaj kaj pli facile efektivigeblaj je komerca skalo kaj sekve, estas la preferata metodo por la produktado de alkilaj poliglikozidoj je granda skalo.
Afiŝtempo: 12 septembro 2020